

Mukopolisaharidoza je vrsta genetsko določenih (pogojenih) patologij, ki se pojavijo v primeru okvarjene presnove kislih mukopolisaharidov (glikozaminoglikanov)..
Najpogosteje je ob taki kršitvi prizadet okostje in zakasnjen fizični razvoj, nekatere vrste patologije pa se kažejo v zaostalosti intelektualne sfere. Poleg tega obstajajo napake v kardiovaskularnem, gastrointestinalnem in živčnem sistemu, optičnih napravah in koži..
Zdravljenje mukopolisaharidoze je simptomatsko, napoved je na splošno neugodna - okvare napredujejo, ker se glukozaminoglikani kopičijo. Bolniki umrejo dovolj zgodaj.
Splošni podatki
Mukopolisaharidozo so preučevali pred kratkim - v 20. stoletju. Sprva je bila obravnavana kot ena patologija, nato pa so zaradi raznolikosti začeli razlikovati njene vrste. Enotni dejavnik vsega mukopolisaharidoze je, da se kisli mukopolisaharidi kopičijo v organih in tkivih, vzrok za to pa je manjvrednost nekaterih celičnih encimov, ki so vključeni v genotip in so podedovani.
Bodite pozorniOrtopedi zdravijo bolnike z mukopolisaharidozo, vendar zahtevajo tudi klinična prizadevanja kardiologov, oftalmologov, nevrologov, otolaringologov in nekaterih drugih strokovnjakov..
Mukopolisaharidoza tipa IH
Mukopolisaharidozo tipa IH imenujemo tudi Hurlerov sindrom. Diagnosticiran je pri 1 dojenčku za 20-25 tisoč.
Prvi znaki bolezni se začnejo pojavljati v prvem letu otrokovega življenja, vendar se lahko popolna klinična slika razvije le 2 leti..
Najbolj značilni simptomi bolezni so:
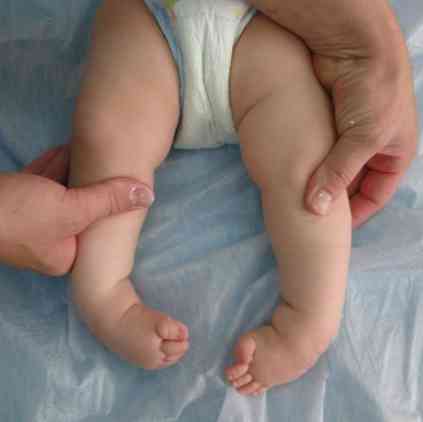 grobi obrisi obraza - zelo značilni za to patologijo;
grobi obrisi obraza - zelo značilni za to patologijo;- deformacija lobanje - postane kot kobilica. To stanje se imenuje scaracephalia;
- dihanje poteka skozi usta, ker je poslabšanje nosnega dihanja zaradi povečanja števila adenoidov, kot tudi nepravilnosti ploda v obrazni lobanji;
- deformacije okončin in nekaterih drugih fragmentov okostja;
- zaviranje rasti;
- ukrivljenost hrbtenice (simptom "mačji hrbet" v sedečem položaju);
- krajšanje vratu;
- netipična visoka razporeditev lopatic;
- izbočena prednja spodnja rebra;
- povečanje širine ščetk - medtem ko so podobne kremplje s krempljem;
- razvoj kontraktur (zmanjšanje obsega gibanja) s postopnim umikom različnih sklepov v patološki proces. Najprej prizadenejo komolce in ramenske sklepe, nato pa se postopek razširi na kolena, kolka in gleženjske sklepe.
 grobi obrisi obraza - zelo značilni za to patologijo;
grobi obrisi obraza - zelo značilni za to patologijo;Za Hurlerjevega sindroma je značilen določen hod - na prstih z ukrivljenimi nogami. Takšna značilnost nastane zaradi omejevanja gibanja zaradi kontraktur..
Fizični pregled razkriva naslednje:
- trebuh je povečan, njegova sprednja stena pri takih bolnikih je oslabljena, zato se lahko pogosto razvijejo popkovna in dimeljska kila;
- povečanje jeter in vranice;
- pri moških se pogosto ugotovi hidrokela - vodeno testis;
- v nekaterih primerih so odkrite kršitve koordinacije gibov;
- pokazala prekomerno rast dlakavih las.
Ko se instrumentalni pregled določi z naslednjim:
- na elektrokardiogramu - znaki razpršene (razširjene) poškodbe srčne mišice;
- okvare vida in sluha;
- oftalmoskopija razkriva motnje in povečanje roženice.
Najbolj značilne so spremembe tega stanja med rentgenskim pregledom:
- X-ray od hrbtenice - vam omogoča, da ugotovite značilno deformacijo hrbtenice. V tem primeru imajo vretenca obliko kocke, njihovi robovi so zaobljeni. Označena je tudi kifoza - ukrivljenost hrbtenice naprej;
- rentgensko slikanje prsnega koša - izrazito debelost prednjih fragmentov reber s hkratnim redčenjem njihovih posteriornih delov, skrajšanjem in deformacijo. Rebra istočasno dobita specifičen videz in postanejo podobna lopati ali sablji. Opažamo tudi zmanjšanje in nekoliko premikanje glavic nadlahtnice;
- radiografija medenice - razkriva zoženje medeničnega obroča, acetabularne votline (tiste, v katerih je "osnova" nastanka kolčnega sklepa) so poševne, glave stegnenice pa zmanjšane;
- Zabeleži se rentgenski pregled tubularnih kosti - redčenje kortikalnega (površinskega) sloja kosti in nekaj ekspanzije medularnega kanala;
- Rentgenski pregled kosti roke - pomaga pri odkrivanju nerazvitosti distalnih (nohtnih) prstnih prstov, skrajšanja in širjenja srednje in proksimalne (tiste bližje dlani) falang, kot tudi metakarpalnih kosti;
- Rentgenska slika lobanje - jasno nerazvite kosti obrazne lobanje, kraniostenoza (zožitev lobanje na nekaterih mestih) in makrocefalija (povečanje lobanje).
Pri bolnikih z diagnozo mukopolisaharidoze tipa IH se ugotovijo naslednje motnje, ki se lahko obravnavajo kot ločene bolezni:
- retinalna pigmentna distrofija - kršitev njenega razvoja;
- glavkom - povečanje intraokularnega tlaka;
- zastoj v fundusu;
- pareza - motnja gibanja;
- paraliza - popolna motnja motorične aktivnosti na določenih področjih mišično-skeletnega sistema.
Sčasoma bolniki z diagnozo Hurler razvijejo in nepovratno napredujejo duševne zaostalosti.
Mukopolisaharidoza tipa I-S
Drugo ime za mukopolisaharidozo tipa I-S je bolezen bolezni. Bolezen je bila opisana šele sredi 20. stoletja, čeprav so se v praksi pred kliničnim zdravnikom pojavili bolniki z značilnimi kontrakturami prstov rok, motnjami gibanja v velikih sklepih in posebnostmi obraza. Simptomi se lahko kombinirajo z znaki Hurlerjevega sindroma.
Ta vrsta mukopolisaharidoze se pojavi pozneje kot pri mukopolisaharidozi tipa IH, njen potek pa je ugodnejši..
Značilnost mukopolisaharidoze tipa I-S je, da do 3-6 let starosti razvoj otrok ustreza starostni normi. Poleg tega se začnejo pojavljati naslednji znaki:
- upogibne kontrakcije prstov;
- omejitev podaljšanja pri velikih in srednje velikih sklepih - namreč, komolca, ramen in zapestja.
Kršitev amplitude gibov na spodnjih udih je manj izrazita kot pri drugih vrstah opisane patologije.
Popolna klinična slika te vrste mukopolisaharidoze je ugotovljena do začetka adolescence.
Fiziološka študija (pregled) upošteva naslednje:
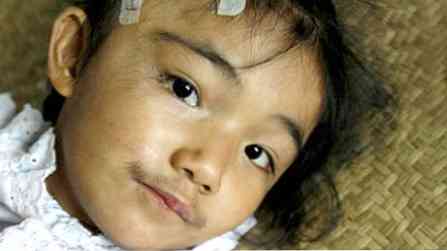 pacienti niso visoki, vendar imajo močno gradnjo (nabito);
pacienti niso visoki, vendar imajo močno gradnjo (nabito);- značilnosti so precej grobe;
- dobro razvita skeletna muskulatura;
- hipertrihoza (povečana telesna dlaka);
- koža na prstih rok (pogosteje) in stopala (manj pogosto) je zgoščena in raztegnjena;
- velikost jeter in vranice se praviloma ni spremenila.
 pacienti niso visoki, vendar imajo močno gradnjo (nabito);
pacienti niso visoki, vendar imajo močno gradnjo (nabito);Poleg tega so pri teh bolnikih ugotovljene kršitve, ki se lahko obravnavajo kot ločena bolezen:
- dimeljska ali popkovna kila;
- sindrom karpalnega tunela - razvija se zaradi izrazite stiskanja mediana živca. Manifestira se z atrofijo (hipoplazijo) tenarskih mišic (dvig v predelu palca) in parestezijami (motnjami občutljivosti) v območju 3, 4 in 5 prstov;
- aortna stenoza - zoženje aorte;
- aortna insuficienca;
- pigmentna distrofija retine;
- glavkom;
- zamračenje roženice.
Inteligentnost bolnikov z diagnozo mukopolisaharidoze tipa I-S ostaja normalna, v nasprotju z bolniki z mukopolisaharidozo tipa IH.
Od instrumentalnih metod je najbolj informativna radiografija. Ugotavlja spremembe, ki so podobne spremembam pri mukopolisaharidozi tipa IH, vendar so manj izrazite..
Mukopolisaharidoza tipa II
Ta tip mukopolisaharidoze imenujemo tudi Hunterjeva bolezen. Patologija najpogosteje prizadene dečke, stare 2-3 leta.
Glede na klinične manifestacije je bolezen podobna mukopolisaharidozi tipa IH - medtem ko so:
- scariocephaly;
- grobe lastnosti;
- nizek ton;
- težave z dihanjem - zaradi deformacije kosti obraznega skeleta.
Z nadaljnjim napredovanjem bolezni se pridružijo naslednji znaki:
- zmanjšana inteligenca;
- koordinacija premikov je prekinjena;
- V takšnem pacientu je razpoloženje nestabilno - opazimo tako imenovano čustveno labilnost - od nasmeha do joka. Takšna nihanja razpoloženja se lahko pojavijo kadarkoli..
Z napredovanjem mukopolisaharidoze tipa II se pojavi in poveča neupravičena agresivnost.
Za razliko od mukopolisaharidoze tipa IH, kršitve hrbtenice v obliki kifoze niso značilne, simptom "mačke nazaj" ni opaziti \ t.
Fizični pregled določa naslednje:
- odkrito je rahlo povečanje vranice in jeter;
- na koži hrbta se določijo specifični vozlički.
Ko je instrumentalni pregled pokazal:
- motnost roženice, ki napreduje;
- povečanje izgube sluha.
Od vseh instrumentalnih metod diagnostike je najbolj informativna radiografija različnih fragmentov mišično-skeletnega sistema - lobanje, celice rude, medenice in tako naprej. Istočasno so radiološki znaki enaki kot pri mukopolisaharidozi tipa I-S.
Bolniki s takšno diagnozo so nagnjeni k razvoju bolezni dihal:
- traheitis;
- bronhitis;
- pljučnica.
Treba je opozoriti, da se mukopopolisaharidoza tipa II lahko razvije v obliki dveh možnosti:
- neugodno (možnost A);
- ugodno (možnost B).
 Neželena varianta se kaže v svetli klinični sliki, hkrati pa so opazne bistvene motnje inteligence. Do smrtnih žrtev lahko pride v adolescenci.
Neželena varianta se kaže v svetli klinični sliki, hkrati pa so opazne bistvene motnje inteligence. Do smrtnih žrtev lahko pride v adolescenci.
Simptomi z ugodno varianto, ki niso izraženi, zgornje kršitve lahko spremlja rahla duševna zaostalost. Takšni bolniki praviloma živijo do 30 let, v nekaterih primerih - in še več.
Mukopolisaharidoza tipa III
Mukopolisaharidozo tipa III je opisal ameriški pediater Sanfillipo, zato je patologija dobila drugo ime - bolezen Sanfillipo.
Bolezen je diagnosticirana pri 1 otroku na 100–200 tisoč novorojenčkov..
Otroci z mukopolisaharidozo tipa III se v prvih letih življenja razvijejo v skladu s starostnimi normami, opazijo pa se lahko tudi nekateri znaki patologije - zlasti težave pri požiranju in neroden hod..
Od 3-5 let, otrok začne zaostajati za vrstniki pri fizičnem razvoju, hkrati pa postane apatičen, brezbrižen do ljudi in dogodkov.. Značilni znaki so:
- motnje govora;
- hrapavost lastnosti;
- redno urinsko inkontinenco (ne le ponoči) in iztrebke.
Ko otrok odraste, se duševna zaostalost poveča in sčasoma postane glavni simptom te vrste mukopolisaharidoze..
Fizikalni pregled je pokazal:
- neznatno povečanje jeter in vranice;
- enakomerno povečanje porazdelitve las;
- kontraktura;
- zaviranje rasti.
 Kršitve organa vida in srčno-žilnega sistema, ki se pogosto pojavljajo pri drugih oblikah opisane patologije, za ta tip mukopolisaharidoze niso značilne.
Kršitve organa vida in srčno-žilnega sistema, ki se pogosto pojavljajo pri drugih oblikah opisane patologije, za ta tip mukopolisaharidoze niso značilne.
Rezultati rentgenskega pregleda z opisano patologijo ali brez patoloških sprememb ali podobni tistim, ki so odkriti pri mukopolisaharidozi tipa I-S, vendar manj izraziti.
Prognoza za to bolezen je manj ugodna kot pri drugih vrstah mukopolisaharlaze - taki bolniki umirajo v starosti od 10 do 20 let, infekcijski zapleti postanejo vzrok smrti..
Mukopolisaharidoza tipa IV
Mukopolisaharidozo tipa IV imenujemo tudi bolezen Morkio (poimenovana po urugvajskem pediatru, ki ga je opisal). Patologija je pogostejša kot pri drugih vrstah mukopolisaharidoze, in sicer pri 1 otroku na 40 tisoč novorojenčkov.
Otroci s to boleznijo do 1-3 let se razvijejo brez posebnosti. Potem pa se pojavijo naslednje kršitve:
- otrok bistveno zaostaja za vrstniki;
- v procesu razvoja je opazil skrajšanje vratu in trupa;
- razvijajo se različne kontrakture;
- koža se zgosti;
- zmanjšuje se mišična moč;
- sluh se slabša;
- obrazne poteze postanejo grobe;
- pojavijo se različne deformacije prsnega koša.
Tudi za mukopolisaharidozo tipa IV obstajajo kršitve, ki se lahko obravnavajo kot ločene patologije:
- valgusova deformacija okončin - ukrivljenost v smeri navzven;
- skolioza (ukrivljenost hrbtenice v lateralni ravnini) ali kifoza;
- dimeljska kila;
- popkovna kila;
- distrofijo roženice.
Razum s shranjeno obliko mukopolisaharidoze.
Diagnozo opisane patologije potrjuje rentgenska slika, ki je v tem primeru najbolj informativna. To so naslednje vrste:
- Rentgenska slika hrbtenice - slike prikazujejo ukrivljenost hrbtenice v obliki kifoze in skolioze ter širjenje in sploščenje hrbteničnih teles;
- radiografija medenice - razkrila je večkratne deformacije obroča medenične kosti, neenakomernost njegovih kontur;
- Detektorji udov - sploščena je glava stegnenice, zaznajo izrazno krajšanje kosti podlaket in deformiranost stopalskih kosti..
Bolniki z mukopolisaharidozo tipa IV živijo praviloma manj kot 20 let. Usodni izid se pojavi na podlagi številnih povezanih bolezni, ki so na koncu zapletene zaradi kardiopulmonalne insuficience..
Mukopolisaharidoza tipa VI
Drugo ime za mukopolisaharidozo tipa VI je Maroto-Lamyjeva bolezen (po imenih dveh francoskih zdravnikov, ki so opisali to patologijo)..
Klinični znaki so večinoma "odmevali" s simptomi drugih vrst mukopolisaharidoze, najbolj izraziti pa so:
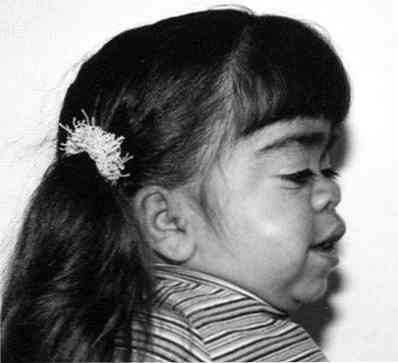 znatno povečanje lastnosti obraza;
znatno povečanje lastnosti obraza;- opazen zaostanek v rasti;
- skrajšanje vratu - včasih se zdi, da bolnikova glava "sedi" skoraj na ramenski pas;
- deformacija prsnega koša.
 znatno povečanje lastnosti obraza;
znatno povečanje lastnosti obraza;Takšni bolniki pogosto imajo prehlade - ta simptom omogoča sum na to posebno in ne na drugo obliko mukopolisaharidoze..
Fizični pregled razkriva naslednje:
- jetra in vranica sta pogosto povečana;
- diagnozo dimeljske kile.
Intelektualno se otrok razvija brez kršitev - ima sposobnost razmišljanja in reševanja intelektualnih problemov..
Obstajata dve različici te vrste mukopolisaharidoze:
- classic;
- mukopolisaharidoza z manjšimi kliničnimi manifestacijami.
Za potrditev diagnoze bo pomagal rentgenski pregled - slike določajo:
- deformacija vretenc - postanejo kvadrati ali klinaste oblike;
- trikotna deformacija medeničnega obroča;
- hipoplazija (nerazvitost) in deformacija glave stegnenice;
- skrajšanje fibule.
Mukopolisaharidozi tipa VII in VIII
 Mukopolisaharidoza teh vrst je manj pogosta od drugih vrst opisane patologije..
Mukopolisaharidoza teh vrst je manj pogosta od drugih vrst opisane patologije..
Mukopolisaharidoza tipa VII se klinično pojavi kot bolezen tipa III, razlika med njimi pa je ugotovljena le v rezultatih biokemičnih študij.
Mukopolisaharidoza tipa VIII je simptomatska in podobna mukopolisaharidozi tipa IV. Vendar pa obstaja razlika v manifestacijah - to je kršitev intelektualnega razvoja, ki ga ne opazimo pri mukopolisaharidozi tipa IV..
Diagnostika
Pri postavitvi diagnoze mukopolisaharidoze temeljijo na pritožbah, anamnestičnih podatkih, rezultatih dodatnih preiskovalnih metod - fizičnem, instrumentalnem, laboratorijskem. Lahko se domneva prisotnost mukopolisaharidoze zaradi značilnih deformacij okostja, ki "ujamejo oči", vendar je možno določiti vrsto opisane bolezni le s pomočjo dodatnih metod preiskave.. Glavne metode so:
- radiografija;
- odkrivanje glikozaminoglikanov v urinu;
- določanje encimske aktivnosti v celičnih kulturah.
Klinični znaki motenj notranjih organov vključujejo metode pregleda:
- elektrokardiografija;
- ultrazvok jeter in vranice;
- elektroencefalografija;
- biokemični test krvi
in mnogi drugi.
Diferencialna diagnostika
Med različnimi tipi mukopolisaharidoze se izvaja diferencialna (značilna) diagnoza.
Zapleti
Najpogostejši zapleti mukopolisaharidoze so:
- kontraktura;
- srčno popuščanje;
- pljučno insuficienco.
Zdravljenje
Zdravljenje mukopolisaharidoze, ki se lahko uporablja za odpravo bolezni, in ne samo njenih manifestacij, ni bilo razvito.. Takšni bolniki prejemajo simptomatsko zdravljenje - lahko je:
- konzervativna - transfuzija krvi, hormonska terapija, vitaminska terapija itd.
- operativno - endoprotetiko glave stegnenice, popravilo kile, kirurški poseg za odpravo kontraktur in popravljanje okostnih deformacij in drugih.
Pomembni so:
- zdravljenje okužb dihalnega trakta;
- popravek vidnega in sluha.
Preprečevanje
Ker patologija nastane zaradi genetskih motenj, ni posebnih preventivnih metod.. Toda tveganje za genske motnje, ki lahko vodijo do razvoja različnih vrst mukopolisaharidoze, se lahko zmanjša, če je ženska dobila normalne pogoje nosečnosti. To je:
- zavračanje slabih navad;
- spoštovanje dela, počitek, spanje;
- optimistično razpoloženje.
Napoved

Prognoza za mukopolisaharidozo je na splošno neugodna, to velja za vse vrste opisanih bolezni. V procesu življenja in razvoja v tkivih se postopoma kopičijo presnovni produkti, ki nastanejo zaradi pomanjkanja tkivnih encimov. To vodi do patoloških sprememb pri skoraj vseh organih in sistemih. Če se začne patologija, se smrt lahko pojavi ne samo zaradi kardiopulmonalne, ampak tudi polisistemske odpovedi.
Konzervativna terapija igra vlogo podpore - uporaba kakršnih koli zdravil vodi v začasno izboljšanje, vendar ne more nadomestiti pomanjkanja tkivnih encimov..
Kovtonyuk Oksana Vladimirovna, zdravstveni komentator, kirurg, svetovalni zdravnik